
FDA对医疗器械的管理通过器械与放射健康中心(CDRH)进行的,中心监督医疗器械的生产、包装、经销商遵守法律下进行经营活动。
医疗器械范围很广,小到医用手套,大至心脏起博器,均在FDA监督之下,根据医疗用途和对人体可能的伤害,FDA将医疗器械分为Ⅰ、Ⅱ、Ⅲ类,越高类别监督越多.
如果产品是市场上不曾存在的新颖发明,FDA要求厂家进行严格的人体实验,并有令人信服的医学与统计学证据说明产品的有效性和安全性。
我们帮助您获得医疗器械的FDA认证,包括:
厂家在FDA注册
产品的FDA登记
产品上市登记(510表登记),产品上市审核批准(PMA审核) 医疗保健器械的标签与技术改造、通关、登记、上市前报告,须提交以下材料:
(1)包装完整的产成品五份,
(2)器械构造图及其文字说明,
(3)器械的性能及工作原理;
(4)器械的安全性论证或试验材料,
(5)制造工艺简介,
(6)临床试验总结,
(7)产品说明书.
如该器械具有放射性能或释放放射性物质,必须详细描述.
所谓GMP就是Good Manufacturing Practice,从字面来看,可以解释成优良制造规范,早在1978年,也就是医疗器材管理法案在美国国会通过之后的第2年,美国就公布实施了医疗器材GMP,从品质管理的理念来看,美国很早就采用了「产品品质是由制造过程所决定」的理念,在当时可算得上**的观念,尤其是将GMP纳入法律之中更可见科技管理法律之进步。
我们先翻一翻法律:在FD&C Act 第520(F)节中规定,医疗器材制造商的设计、制造、包装、储存及组装的过程、设施及品质管制等过程都必须符合一般性要求,这些要求包括下列:
1. 组织与员工;
2. 建筑物、设备、对零组件、制程、包装、标示的管制;
3. 器材的组装、销售及仓储;
4. 器材功能及安全性的评估;
5. 器材与制程的记录、抱怨及
6. 品质系统稽核。
这些要求的具体实践,就是制造商的品质保证系统,与近10年来蔚为风潮的ISO 9000品质系统相符合,不过FDA的GMP查厂实务与ISO 9000指定机构(Notified Body)的实务有很大的不同,除了FDA公告免除GMP的产品以外(部份Class I的器材不需要实施GMP,其名单可见于21CFR 882.1525),所有制造商一律要实施GMP,FDA的查厂政策是选择性的抽查,查厂完成并不给予证书,请读者特别注意到这些差异。当然FDA的GMP与ISO 9001, ISO 13485及EN 46001有许多相同与相异之处。
历经20年的实施,GMP也一再修正,主要的修正从1990年开始,如医疗器材安全法SMDA修正案,就增加了对设计验证、器材安全与功效性以外的性能评估要求。1996年更公布了-新修正的GMP并更名为品质系统规范(Quality System Regulation, QSR),1997年6月起正式实施,这份新规范采用了ISO 9001的架构,除了原有的GMP精神之外,更加强了设计管制及计算机软件验证的要求。
美国有关医疗器材管理的法规美国有关医疗器材管理的法规主要有下列三项:
(1)1938年的联邦食品药物及化妆品法(The Federal Food, Drug &
Cosmetic Act of 1038, 简称FD&C Act of 1938)。此法案在1938年之后
有三次重大修正,分别为:
(A)1976年医疗器材修正案(The Medical Device Amendments of1976, 简称the 1976 Amendments)
(B)1990年安全医疗器材法案(The Safe Medical Devices Act of1990, SMDA)
(C)1992年医疗器材修正案(The Medical Devices Amendments of1992, 简称the 1992 Amendments)
(2)公共保健服务法(Public Health Service Act)
(3)包装与标示法(Fair Packaging and Labeling Act)
 2022年-中铁二十局集团有限公司雄商高铁站前七标项目经理部
2022年-中铁二十局集团有限公司雄商高铁站前七标项目经理部 2022年-陕西延长石油延安能源化工有限责任公司聚烯烃灌装
2022年-陕西延长石油延安能源化工有限责任公司聚烯烃灌装 2022年-麻家梁煤业有限责任公司工作面奥灰水地面区域治理
2022年-麻家梁煤业有限责任公司工作面奥灰水地面区域治理 2022年-亚洲基础设施投资银行贷款河南郑州等地特大暴
2022年-亚洲基础设施投资银行贷款河南郑州等地特大暴
 华润电力红安天明150MW风电项目220kV升压站PC工程
华润电力红安天明150MW风电项目220kV升压站PC工程 华润清远清新林泉扩建50MW风电项目主体施工工程招标公告
华润清远清新林泉扩建50MW风电项目主体施工工程招标公告 华润水泥合浦分布式光伏项目EPC工程总承包招标公告
华润水泥合浦分布式光伏项目EPC工程总承包招标公告 华润电力鲤鱼江电厂贮灰场环境治理及综合利用光伏项目110kV
华润电力鲤鱼江电厂贮灰场环境治理及综合利用光伏项目110kV 山西阳泉矿区泊里煤矿项目井底车场水仓、主排水泵房及主变电所掘
山西阳泉矿区泊里煤矿项目井底车场水仓、主排水泵房及主变电所掘 山西乡宁焦煤集团东沟煤业有限公司工业广场边坡项目治理工程总
山西乡宁焦煤集团东沟煤业有限公司工业广场边坡项目治理工程总 利用亚行贷款中国—东盟中小企业协同创新发展综合提升
利用亚行贷款中国—东盟中小企业协同创新发展综合提升 中铁二十局集团有限公司雄商高铁站前七标项目经理部粉煤灰
中铁二十局集团有限公司雄商高铁站前七标项目经理部粉煤灰 陕西延长石油延安能源化工有限责任公司聚烯烃灌装改造
陕西延长石油延安能源化工有限责任公司聚烯烃灌装改造 麻家梁煤业有限责任公司工作面奥灰水地面区域治理工程招标公告
麻家梁煤业有限责任公司工作面奥灰水地面区域治理工程招标公告 亚洲基础设施投资银行贷款河南郑州等地特大暴雨洪涝灾害灾后恢复
亚洲基础设施投资银行贷款河南郑州等地特大暴雨洪涝灾害灾后恢复 (2022年)鲁山豫能抽水蓄能有限公司河南鲁山抽水蓄能电站安
(2022年)鲁山豫能抽水蓄能有限公司河南鲁山抽水蓄能电站安 (2022年)海南新媒体绿都一期项目(西地块)电梯采购与安装
(2022年)海南新媒体绿都一期项目(西地块)电梯采购与安装 (2022年)同煤大唐塔山煤矿有限公司四盘区立井井筒装备安装
(2022年)同煤大唐塔山煤矿有限公司四盘区立井井筒装备安装 (2022年)亚洲开发银行贷款山西城乡水源保护和环境改善示范
(2022年)亚洲开发银行贷款山西城乡水源保护和环境改善示范 2022年-首钢股份公司迁安钢铁公司炼铁作业部烧结精
2022年-首钢股份公司迁安钢铁公司炼铁作业部烧结精 2022年-江苏省液化天然气储运调峰工程项目取排水工程施工
2022年-江苏省液化天然气储运调峰工程项目取排水工程施工 2022年-山西忻州神达万鑫安平煤业有限公司矿井兼并重组整合
2022年-山西忻州神达万鑫安平煤业有限公司矿井兼并重组整合 2022年-山西忻州神达原宁煤业有限公司90万吨/年矿井兼并
2022年-山西忻州神达原宁煤业有限公司90万吨/年矿井兼并 2022年-成庄矿选煤厂煤泥干燥系统改造工程招标公告
2022年-成庄矿选煤厂煤泥干燥系统改造工程招标公告 2022年-高青县春汇综合智能仓配物流园项目施工总承包
2022年-高青县春汇综合智能仓配物流园项目施工总承包 2022年-陕西中烟工业有限责任公司汉中卷烟厂卷包除尘
2022年-陕西中烟工业有限责任公司汉中卷烟厂卷包除尘 2022年-同煤大唐塔山煤矿有限公司煤泥烘干生产线清洁热源工
2022年-同煤大唐塔山煤矿有限公司煤泥烘干生产线清洁热源工 2022年-承德航天天启风光储氢一体化多能互补示范项目
2022年-承德航天天启风光储氢一体化多能互补示范项目 2022年-利用亚洲开发银行贷款农业综合开发长江绿色生态廊道
2022年-利用亚洲开发银行贷款农业综合开发长江绿色生态廊道 2022年-嵩县前河矿业有限责任公司葚沟矿区天井钻机工程
2022年-嵩县前河矿业有限责任公司葚沟矿区天井钻机工程 2022年-广东陆河抽水蓄能电站施工电源工程建设项目招标公告
2022年-广东陆河抽水蓄能电站施工电源工程建设项目招标公告 首钢股份公司迁安钢铁公司炼铁作业部烧结精3、返8通廊加固设计
首钢股份公司迁安钢铁公司炼铁作业部烧结精3、返8通廊加固设计 江苏省液化天然气储运调峰工程项目取排水工程施工招标公告
江苏省液化天然气储运调峰工程项目取排水工程施工招标公告 山西忻州神达万鑫安平煤业有限公司矿井兼并重组整合项目矿建工程
山西忻州神达万鑫安平煤业有限公司矿井兼并重组整合项目矿建工程 山西忻州神达原宁煤业有限公司90万吨/年矿井兼并重组整合项目
山西忻州神达原宁煤业有限公司90万吨/年矿井兼并重组整合项目 旧大梁切割拆除-墙锯切割-水钻切割方法【拆除视频】
旧大梁切割拆除-墙锯切割-水钻切割方法【拆除视频】 河南燃气圆球形爆米花机多少钱一台
河南燃气圆球形爆米花机多少钱一台 中山ITO膜激光打标机,小榄手机外壳激光雕刻机,镭雕机供应
中山ITO膜激光打标机,小榄手机外壳激光雕刻机,镭雕机供应 SD-R16手动热熔打包机
SD-R16手动热熔打包机 变色毛巾批发厂家
变色毛巾批发厂家 消夏凉坐垫
消夏凉坐垫 中诚ERP软件(机械版)演示视频教程8-储备件生产
中诚ERP软件(机械版)演示视频教程8-储备件生产 LED油压剪脚机
LED油压剪脚机 被动片
被动片 公司合作
公司合作 贵州核桃苗,贵州川早2号核桃苗,贵州核桃苗挖坑
贵州核桃苗,贵州川早2号核桃苗,贵州核桃苗挖坑 山东省盛华万头肉牛肉羊牧业牛羊养殖场
山东省盛华万头肉牛肉羊牧业牛羊养殖场 catalogue产品目录
catalogue产品目录 通风管道加工设备
通风管道加工设备 杭州汇毅
杭州汇毅 如皋汽车导航仪GPS终端
如皋汽车导航仪GPS终端 沙溪回收陶瓷电容, 收购EP1AGX20CF484I5N
沙溪回收陶瓷电容, 收购EP1AGX20CF484I5N 废电线电缆回收,废金属回收,旧空调回收,废品回收
废电线电缆回收,废金属回收,旧空调回收,废品回收 西门子 触摸屏 变频器 软件 PRC CPR 模块 回收高价
西门子 触摸屏 变频器 软件 PRC CPR 模块 回收高价 上门采购Agilent4286A HP4286A回收无极限
上门采购Agilent4286A HP4286A回收无极限 回收库存UV油漆
回收库存UV油漆 佳木斯回收五金烤漆13673108955
佳木斯回收五金烤漆13673108955 想询问越秀上门回收洋酒行情`,广州大量回收洋酒
想询问越秀上门回收洋酒行情`,广州大量回收洋酒 临沧哪里回收聚乙烯蜡 蜡片高价回收13931082352
临沧哪里回收聚乙烯蜡 蜡片高价回收13931082352 P91等无缝合金管件最新价格 大量库存 欢迎订购
P91等无缝合金管件最新价格 大量库存 欢迎订购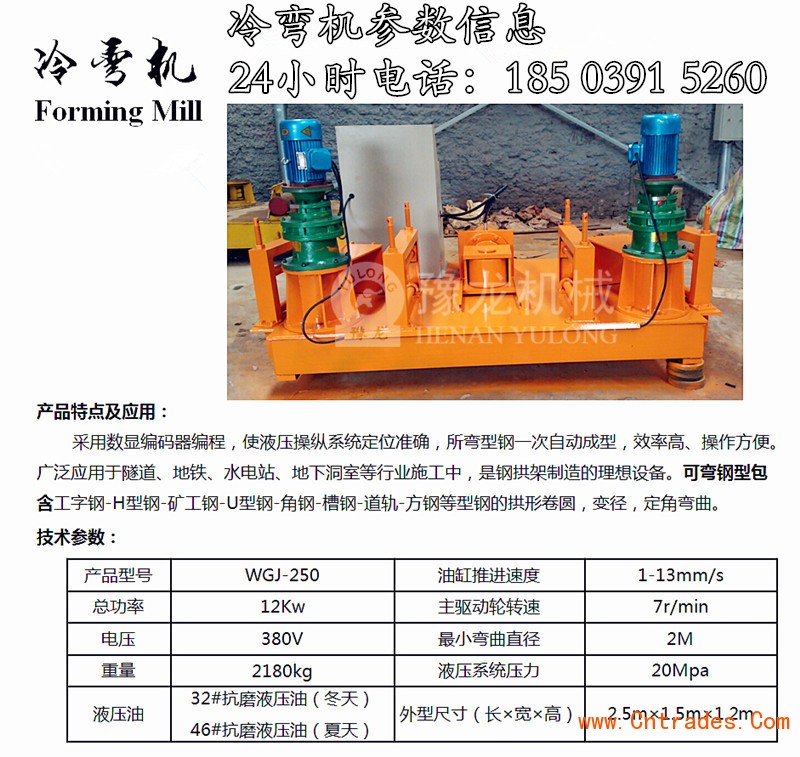 安徽工字钢顶弯机多少钱
安徽工字钢顶弯机多少钱 内蒙古钢绞线穿梭机批发钢绞线自动穿梭机
内蒙古钢绞线穿梭机批发钢绞线自动穿梭机 黄子龙工作室经纪公司13144445077
黄子龙工作室经纪公司13144445077 测量模式:快速筛查,自动抽气式 测量范围:0-100mg/100ml BAC(血液酒精浓度) 测量误差当吹气距离(仪器与嘴之间距离)小于5cm:±5.0mg/100ml 当吹气距离(仪器与嘴之间距离)
测量模式:快速筛查,自动抽气式 测量范围:0-100mg/100ml BAC(血液酒精浓度) 测量误差当吹气距离(仪器与嘴之间距离)小于5cm:±5.0mg/100ml 当吹气距离(仪器与嘴之间距离) 景区售卖花车,室外广场小吃售卖亭
景区售卖花车,室外广场小吃售卖亭 耐用变频器ACS550-01-06A9-4
耐用变频器ACS550-01-06A9-4 E2E-X10D1-M1GJ-Z
E2E-X10D1-M1GJ-Z




")













